Proteomics can be broadly classified into three types namely:
- Expression proteomics,
- Structural proteomics and
- Functional proteomics.
Expression Proteomics
This is the quantitative characterization of protein expression at the whole proteome level. It involves the quantitative measurement of proteins in a cell at a particular metabolic state. Before proceeding for expression analysis, the expressed proteins in a proteome are determined. The proteins are separated, identified and quantified. The comparative profiling of proteins is often performed after separating the proteins by two-dimensional (2D) gel electrophoresis and identifying them by mass spectrometry (MS).
The various steps involved can be outlined as follows:
- Protein separation on 2D gel electrophoresis,
- Protease digestion,
- Mass spectrometry and
- Peptide identification.
Protein separation on 2D gel electrophoresis
The 2D protein gel electrophoresis is a method of protein separation that enables to distinguish up to 10,000 proteins. First, proteins are separated according to their isoelectric point (the pH at which the net charge of the protein equals zero). The proteins are loaded onto a pH gradient and are made to migrate under the influence of an electric field. The protein migrates through the gradient towards anode or cathode until their isoelectric point is reached beyond which their migration stops.
The proteins are then separated by common poly acrylamide gel electrophoresis (PAGE). The electric current is now applied perpendicular to the original orientation of the electrodes. The proteins now migrate through the gel only according to their size. After the 2D electrophoresis of the gel, it is visualized by suitable staining or labelling methods.
The resulting protein profile can be compared, for example, between experimental and control samples. The differentially expressed proteins are identified, cut out from the gel and subjected to subsequent analysis by MS.
Mass spectrometry
This method enables precise measurement of molecular weight of a broad spectrum of substances. As the studied substance has to be intact in gas phase, MS for protein analysis was enabled by the development of ‘soft’ ionization techniques of MS such as matrix-assisted laser detection of desorption/ionization (MALDI) and electrospray ionization (ESI).
Protein identification is generally performed in two ways:
- Protein is digested by trypsin or by other proteolytic enzyme to smaller peptides and their precise molecular weights are measured using MS. The spectrum of the molecular weights is then compared with theoretical spectra that are calculated from protein sequences from available databases (using bioinformatics tools).
- Tandem MS enables to choose the peptide which is then fragmented by the collision with inert gas. The fragmentation pattern gives either full of partial information about protein sequence that is subjected to the search in databases.
MS also helps in protein post-translational modification analysis, because it enables to localize given modifications within the protein and also detects the nature of such modification.
Peptide identification
Once the peptide mass finger prints or peptide sequences are determined, bioinformatics programmes can be used to search for the identity of a protein in a database of theoretically digested proteins. For example, ExPASY (www.expasy.ch/tools/) is a proteomics web server with programs for searching peptide information from the protein databases such as SWISS-PROT. Mascot (www.matrixscience.com/search_form_select.html) is another web server that identifies proteins based on peptide mass finger prints and also sequences entries.
Structural Proteomics
This involves the determination of the 3D structure of proteins. Structural proteomics identifies all the proteins within an organelle, determines their locations and characterizes their interactions.
Post-translational modifications play a very important role in proteome analysis. These modifications have a great impact on protein function by altering the size, hydrophobicity and overall conformation of the proteins. Further, the modifications can directly influence the protein-protein interaction and the distribution of proteins to different subcellular locations. Various bioinformatics tools predict sites for post-translational modifications based on specific protein sequences. To minimize false positive results, a statistical process called support vector machine can be used and this increases the specificity of such predictions. AutoMotif (http://automotif.bioinfo.pl/) is a web server for predicting protein sequence motifs.
Some of the other sites for online analysis of proteins structures are:
- TMpred: Prediction of Trans-membrane Regions and Orientation (http://www.ch.embnet.org/software/TMPRED_form.html).
- TMHMM: Prediction of transmembrane helices in proteins
(http://www.cbs.dtu.dk/services/TMHMM-2.0/). - DAS: Transmembrane Prediction Server (http://www.sbc.su.se/~miklos/DAS/).
- SPLIT: The Trans-membrane Protein Topology Prediction Server provides clear and colourful output including beta preference and modified hydrophobic moment index. (http://split.pmfst.hr/split/4/).
- OCTOPUS: Predicts the correct topology for 94 percent of the dataset of 124 sequences with known structures. (http://octopus.cbr.su.se/).
- SLEP (Surface Localization Extracellular Protein): For predicting the localization of bacterial proteins starting from genome sequences (http://bl210.caspur.it/slep/slep_newJob.php).
- SignalP: Predicts the presence and location of signal peptide cleavage sites in Gram-positive, Gram-negative and eukaryotic proteins (http://www.cbs.dtu.dk/services/SignalP/).
- pTARGET is a computational method to predict the subcellular localization of only eukaryotic proteins from animal species that include fungi and metazoans. Predictions are carried out based on the occurrence patterns of protein functional domains and the amino acid compositional differences in proteins from different subcellular locations. This method can predict proteins targeted to nine distinct subcellular locations that include cytoplasm, endoplasmic reticulum, extracellular/secreted, Golgi bodies, lysosomes, mitochondria, nucleus, peroxysomes and plasma membrane (http://bioapps.rit.albany.edu/pTARGET/).
Prediction of disulphide bridges
Disulphide bridge is a unique post-translational modification. Disulphide bonds are very much essential for maintaining the stability of proteins. Prediction of disulphide bonds may help to predict the 3D structure of proteins. For example,
DiANNA: a web server for disulphide connectivity prediction. The web server bc.edu/~clotelab/DiANNA/ outputs the disulphide connectivity prediction given input of a protein sequence.
DBCP: a web server for disulphide bonding connectivity pattern prediction without the prior knowledge of the bonding state of cysteines.
Prediction of protein-protein interactions
Proteins should interact with each other to carry out biochemical functions. Thus, the prediction of protein-protein interactions is an important aspect of proteomics. A number of computational approaches have been developed for the prediction of protein-protein interactions. These methods utilize the structural, genomic and biological contexts of proteins and genes in complete genomes to predict protein interaction networks and functional linkages between proteins. STRING is a database of known and predicted protein interactions. The interactions include direct (physical) and indirect (functional) associations; they are derived from four sources: namely genomic, high-throughput, conserved/co-expression and previous knowledge.
Figure 11.7 Phylogenetic profile
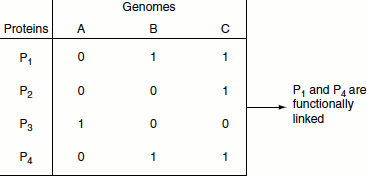
Protein-protein interactions can be predicted by different methods, namely:
- Gene co-expression,
- Gene cluster and gene neighbour,
- Phylogenetic profile (Figure 11.7),
- Rosetta stone,
- Bayesian networks,
- Sequence evolution and
- Random decision forests.
Rosetta stone
This method is based on gene events. If A and B exist as interacting domains in a fusion protein in one proteome, the gene encoding the protein is a fusion gene. Their homologous gene sequences A’ and B’, which exist separately in another genome, most likely encode proteins interacting to perform a common function. On the other hand, if ancestral genes A and B encode interacting proteins, they may have a tendency to be fused together in other genomes during evolution to enhance their functionality. This method of predicting protein-protein interactions is called the Rosetta stone (Figure 11.8).
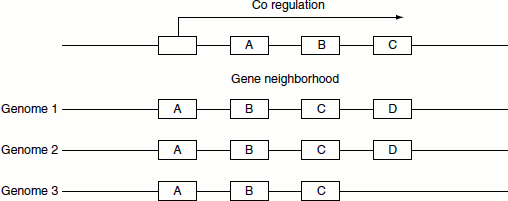
Gene cluster and gene neighbour
If certain gene linkage is found to be conserved across divergent genomes, it can be used as a strong indicator of formation of an operon. This method of prediction is better valid for prokaryotes; however, in eukaryotes, gene order is a less potent predictor of protein interactions (Figure 11.9).
Figure 11.8 Rosetta stone
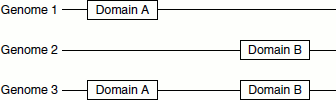
Figure 11.9 Gene cluster
Functional Proteomics
Functional proteomics is an emerging research area that focuses on the identification of biological functions of unknown proteins and also defines the cellular mechanisms at the molecular level. Understanding protein functions as well as unraveling molecular mechanisms within the cell depend on the identification of the interacting protein partners. The association of an unknown protein with partners belonging to a specific protein complex involved in a particular mechanism would be strongly suggestive of its biological function. Such protein–protein interaction studies also details the cellular signaling pathways. Functional proteomics can define prognosis and predict pathologic complete response in patients and hence is more appropriately referred as ‘clinical proteomics’. A variety of MS-based approaches allow the characterization of cellular protein assemblies under near-physiological conditions and subsequent assignment of individual proteins to specific molecular machines, pathways and networks, etc.
Protein microarrays (protein chips)
These are similar to DNA microarrays. A large number of proteins can be analysed. These protein chips contain entire immobilized proteome. Unlike in DNA microarray, these are not used to bind and quantitate complementary molecules but are used for studying protein function.
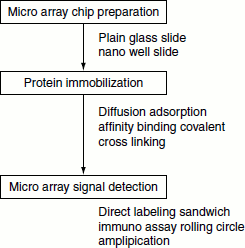
Protein arrays are solid-phase ligand-binding assay systems using immobilized proteins on surfaces which include glass, membranes, microtiter wells, mass spectrometer plates and beads or other particles. There are rapid and automatable, highly sensitive, economical and gives an abundance of data for a single experiment (Figure 11.10).
Types of protein arrays
Protein arrays are of three types namely:
- Large-scale functional chips (target protein arrays): These are constructed by immobilizing a large numbers of purified proteins. This type of protein array is used to assay biochemical functions such as protein–protein, protein–DNA, protein-small molecule interactions and enzyme activity, and to detect antibodies and their specificity.
- The analytical capture arrays: These contain affinity reagents, primarily antibodies. They are used to detect and quantitate analytes in plasma/serum or tissue extracts
- Lysate (reverseprotein) arrays: In this type of array, the complex samples—such as tissue lysates—are coated on the surface and target proteins are then detected with antibodies overlaid on the coated surface.
Figure 11.10 Manufacturing of protein miccroarays
Protein sources
Sources of proteins for the construction of arrays, include cell-based expression systems for recombinant proteins, proteins purified from natural sources, proteins produced in vitro by cell-free translation systems, and peptides prepared by synthetic methods. Many of these methods can be automated for high-throughput production.
Solid surfaces
Protein arrays are basically mini versions of familiar immunoassay methods such as ELISA and dot blotting. They employ the use of fluorescent readout, robotics and high-throughput detection systems. This enables multiple assays to be carried out in parallel. The commonly used physical supports for the protein arrays include glass slides, silicon, microwells, nitrocellulose membranes, magnetic and microbeads. Micro-drops of protein are delivered onto planar surfaces.
Protein immobilization
A good protein array support surface should have the following features:
- It should be chemically stable before and after the coupling procedures.
- It should allow good spot morphology.
- It should display minimal non-specific binding.
- It should not contribute a background in detection systems.
- It must be compatible with different detection systems.
- The immobilization method used should be reproducible.
- It is applicable to proteins of different properties (size, hydrophilic and hydrophobic).
- It is amenable to high throughput and automation.
- It is compatible with the retention of fully functional protein activity.
Proteins are immobilized both covalently and non-covalently. For example, diffusion into porous surfaces (allows non-covalent binding of unmodified protein within hydrogel structure), passive adsorption to surfaces, covalent binding using tags such as biotin/avidin on the protein bind the protein specifically.
They provide a solid support for assaying enzyme activity, protein-protein interaction, protein DNA/RNA interaction, protein ligand interaction, etc. Antibodies can be fixed on a solid support for assaying thousands of proteins simultaneously.
The protein chips thus created, helps to assay enzymes, protein-protein interaction, protein DNA/RNA interaction, protein ligend interaction etc. Antibodies can be fixed on a solid support for assaying thousands of proteins simultaneously.
Applications
- Diagnostics: Detection of antigens and antibodies in blood samples; profiling of sera to discover new disease markers; environment and food monitoring. Also finds applications in autoimmunity, allergy and cancer.
- Proteomics: Protein expression profiling.
- Protein functional analysis: Protein-protein interactions; ligand-binding properties of receptors; enzyme activities.
- Antibody characterization: Cross reactivity and specificity, epitope mapping.