The first step in the rDNA technology is the isolation of the desired gene of interest for which the DNA from the cell has been isolated.
DNA Isolation
DNA can be isolated by employing gentle methods of cell rupture. Cell walls if present are digested enzymatically (lysozyme treatment) and the cell membrane is solubilized using detergent. Autoclaving is necessary to eliminate DNase activity. Once nucleic acids are released from the cell, RNA can be removed by treatment with RNase. The proteins can be removed by treatment with water-saturated phenol or with phenol/chloroform mixture. This denatures the protein but not the nucleic acids. The emulsion formed is centrifuged and the protein precipitated. The aqueous layer is recovered and deproteinized repeatedly. This is followed by centrifugation. The supernatant is recovered and treated with ethanol that precipitates the DNA. The precipitated DNA is recovered and dissolved in a buffer containing EDTA (to eliminate DNAse action) and stored at 4°C. This procedure is best suited for cellular DNA. If DNA from specific organelle or viral particle is needed, it is best to isolate the organelle or virus before extracting the DNA (Figure 9.1).
Figure 9.1 DNA isolation

DNA Sequencing
The nucleotide sequence of the gene of interest can be analysed. There are two main methods of nucleic acid sequencing namely:
- Maxam and Gilbert’s chemical method and
- Sanger’s dideoxy method or enzymatic method or chain termination method.
Maxam and Gilbert’s chemical method
Allan Maxam and Walter Gilbert developed a chemical method for DNA sequencing. However, the method is laborious and time-consuming.
The basic steps employed are:
- Double-stranded DNA is separated.
- The separated single-stranded DNA is end labelled at its 5′-end by using polynucleotide kinase.
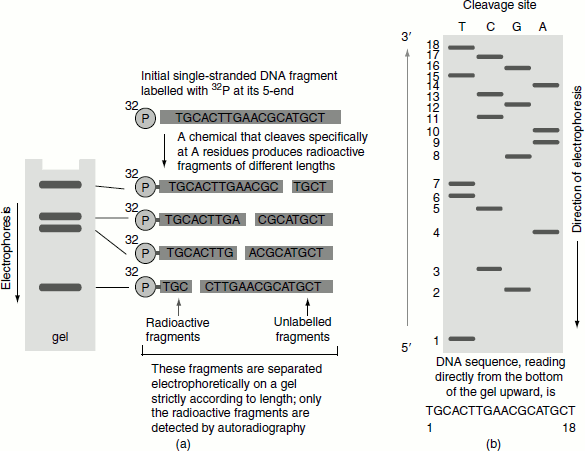
- The DNA strand is exposed to mild treatment with a chemical that destroys one of the four bases (e.g., only A residues). Since the treatment is mild, usually only one of the A residues in each molecule is destroyed at random. As the result, a group of DNA fragments of different lengths, reflecting the different sites at which A residues occur in the original DNA are generated.
- These fragments are separated on a gel and detected by autoradiography and their sizes reveal the distances from the labelled end and the A residues.
- Similar procedures are carried out simultaneously on four separate samples of the same 5′-end-labelled DNA molecule using chemicals that cleave DNA preferentially at T, C and G respectively.
- The resulting fragments are separated on an agarose gel, giving a pattern of radioactive DNA bands from which the DNA sequence is read. The nucleotide closest to the 5′-end of the sequence is determined by looking across the gel at level 1 (at the bottom of the gel) and see-ing in which lane a band appears (T). The same procedure is repeated for level 2, then level 3 and so on, to obtain the sequence (Figure 9.2).
Figure 9.2 Maxam and Gilbert’s chemical method of DNA sequencing
Sanger’s dideoxy method
This method employs the following steps:
- Double-stranded DNA is separated.
- Separated DNA template, DNA polymerase Klenow fragment (the larger domain of DNA polymerase-I (DNA Pol-I), lacking 5′ → 3′ exonuclease activity), labelled primer, deoxyribo-nucleotides and four different dideoxyribonucleotide, each one specially added in one tube, are taken in four reaction tubes (Figure 9.3).
- When such a modified nucleotide is incorporated into a DNA chain, it blocks the addition of the next nucleotide due to the absence of a 3′-OH group.
- Each newly synthesized DNA strand made in a test tube by DNA polymerase will stop at a randomly selected base in the sequence.
- This reaction, therefore, generates fragments of DNA similar to that explained previously for the chemical method. These fragments are detected by a label (chemical or radioactive) that is either incorporated into the oligonucleotide primer or into one of the deoxyribonucleoside triphosphates used to extend the DNA chain.
Figure 9.3 DNA sequencing by Sanger’s dideoxy method
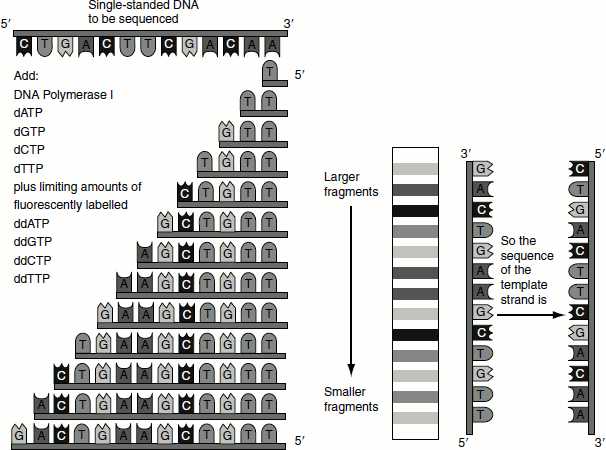
Figure 9.4 Sanger’s dideoxy method of DNA sequencing
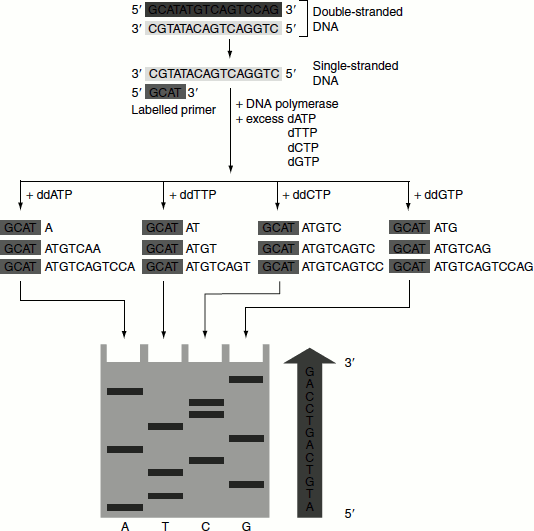
- To determine the full sequence, four different chain-terminating nucleoside triphosphates are used in separate DNA synthesis reactions on the same primed single-stranded DNA template.
- When the products of these four reactions are analysed by electrophoresis in four parallel lanes of a polyacrylamide gel, the DNA sequence can be derived in the same way as explained for the chemical method (Figure 9.4).
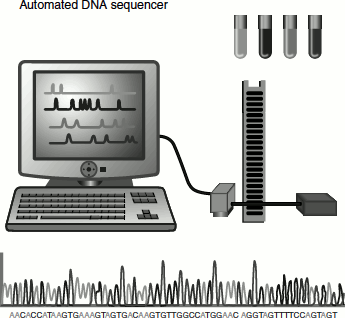
Automated DNA Sequencing
Sanger’s method was automated in 1986 by Leroy Hood and Loyd Smith. In this method, each ddNTP is tagged with a fluorophore of four different colours. Thus, instead of having four separate sequences as described in the previous methods, the reactions can be combined into one tube. The DNA fragments are separated using a polyacrylamide gel. A laser beam excites the flurophore tagged to the fragments as they reach the detector near the end of the gel. These signals are fed to a computer that reads the results as DNA sequence. About 4,800 bases of sequence can be read per day by this method. Currently more automated sequencers are used, which can detect as many as 2 million base sequences per day.
The above picture depicts the steps involved in automated DNA sequencing. The technique uses dideoxynucleotides, just as described in other methods, but the primers used in each of the four reactions are tagged with different fluorescent molecules. The products from each tube will emit a different colour fluorescence when excited by light.