Gel electrophoresis uses electricity to separate charged macromolecules by size, as they migrate through a gel matrix. The gel acts as a molecular sieve, as the macromolecular mixture moves through the gel under the influence of an electric field. Thus, the molecules of varying size are separated.
DNA Electrophoresis
‘Agarose gel electrophoresis’ is the easiest and commonest way of separating and analysing DNA (Figure 12.1). DNA is a negatively charged molecule and is moved by electric current through a matrix of‘agarose’. Agarose is available in powdered form and is insoluble in water or buffer at room temperature; however, it dissolves in boiling water. When the water starts cooling, the process of polymerization takes place, causing the sugar polymers crosslink with each other and forms the gel.
Figure 12.1 Agarose gel electrophoresis
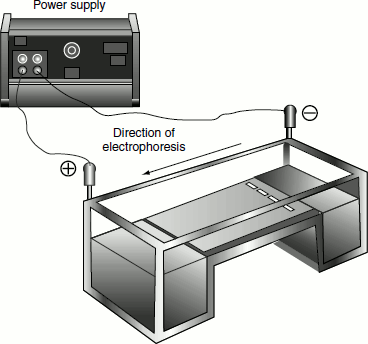
The DNA is forced to migrate through a highly cross-linked agarose matrix in response to an electric current. In solution, the phosphates on the DNA are negatively charged, and the molecule will, therefore, migrate towards the positive electrode (anode).
The rate of DNA migration through the agarose gel is influenced by three factors namely:
- size of the DNA,
- conformation of the DNA and
- ionic strength of the running buffer.
Larger DNA fragments are more easily entangled in the agarose gel matrix and hence they migrate slowly through the gel. Smaller fragments run more quickly than large fragments. The pore size of the gel matrix can be adjusted by varying the concentration of agarose. A standard 1 per cent agarose gel can resolve DNA from 0.2-30 kb in length. Agarose gels are referred to as submarine gels, because the slab is laid horizontally and is completely covered by running buffer.
The various steps underlying agarose gel electrophoresis can be discussed as follows:
Gel preparation: Gels are prepared as percentage weight/volume solutions. A 1 per cent gel is 1 g agarose in 100 ml buffer (Tris Boric acid EDTA buffer, TBE buffer is generally used). The agarose mixed solution is then melted and cooled. Small volume of ethidium bromide can be mixed with the melted agarose. This will enable visualization of the separated DNA fragments. Ethidium bromide is a powerful mutagen and hence need to be handled carefully using gloves.
Gel casting: The gel suitably prepared is then poured into gel trays that are made of UV-transparent plastic and allowed to set after placing the gel comb that forms the wells for loading the sample. The gels are usually made of a thickness of 5–7 mm.
Reservoir buffer: 1X TBE Running Buffer is prepared by mixing water, Tris Base, boric acid and disodium EDTA. The buffer is then added to both reservoirs and the gel is covered with the buffer to a depth of about 2 mm.
Sampling loading: After adding the running buffer, the comb is carefully removed and the DNA sample to be separated is added to the sample well after mixing it with the gel loading buffer. In one of the sample wells, a marker DNA also called ladder DNA can be added, which will enable to identify the molecular size of the sample DNA.
Running the gel: Once the sample is loaded to the gel, electrophoresis is started by connecting to the electrodes. The power pack is then turned on and the gel is run for the appropriate length of time. The gel can run at about 60–80 V for about 40 min. The larger gels can be run at 100–105 V for about 2 h, or at 15 V for an overnight electrophoresis. When the gels are run at greater voltages, it will result in the heating of the gel and the distortion of the bands. The gels run at very high voltage can deliver powerful electric shocks.
Figure 12.2 Agarose gel electrophoretic pattern of a DNA sample
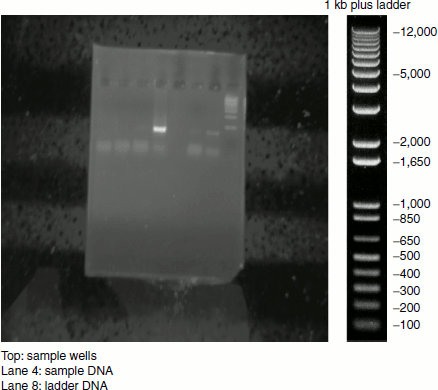
Visualizing the gel: After the run, the power pack is turned off and the electrodes are disconnected. The sample run gel is carefully laid onto a UV transilluminator when the DNA fragments appear as coloured bands (Figure 12.2). The separated sample DNA can be compared with the ladder DNA and can be inferred about its size.
Protein Electrophoresis
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis
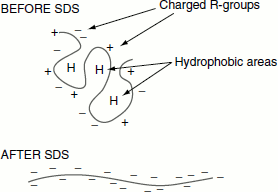
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) is the commonly employed technique for separating proteins. Proteins are separated based on their size. In this method, the protein mixture is sieved through a polyacrylamide gel. The charged protein molecules migrate through the gel under the influence of an electric field. The sample proteins may carry varying charges and may not migrate only according to their size. In order to make the proteins have a uniform charge to mass ratio, the proteins are treated with sodium dodecyl sulphate (SDS), an anionic detergent that denatures the proteins. SDS coats the whole protein molecule. Thus, after SDS treatment, the entire native charge carried by the protein is masked and all proteins become negatively charged and migrate in the gel towards the anode based on their mass only (Figure 12.3). About 1.4 g of SDS binds per gram of the protein.
Figure 12.3 SDS masking the native charge of protein
Gelpreparation: Polyacrylamide is a polymerofacrylamide cross linked by N,N′-methylenebisacrylamide. For the polymerization ammonium persulphate and TEMED (N,N,N′,N′-tetramethyl-ethane-1,2-diamine) are also required (Figure 12.4).
Ammonium persulphate (APS) provides the free radicals that are necessary for the polymerization reaction and TEMED initiates the reaction. Varying percentage of the gel from 5-25 per cent can be prepared by varying the concentration of acrylamide and bisacrylamide.
Gel casting: Two small glass plates are cleaned with acetone/ethanol and are clamped together after spacing the spacers (thin plastic strips that create a thin space between the glass plates). The ends of the glass plates all suitably sealed on all sides except on the top. The gel solution (the mixture of acrylamide, bisacrylamide, APS, TEMED and buffer) is then poured between the glass plates and allowed to set.
Separating gel/resolving gel: The gel solution poured between the gel plates is not uniform throughout, instead it is divided into two portions namely the stacking gel region and the separating gel. These two regions differ in the concentration of acrylamide and bisacrylamide and in pH. The separating gel is a less pore size gel prepared by using increased acrylamide–bisarylamide concentration (10–15 per cent) and has a pH of 8.8 (tris-HCl buffer used). The actual separation of the protein takes place in the separating gel (Figure 12.5).
Figure 12.4 Polymerization of acrylamide and bisacrylamide to form polyacrylamide gel
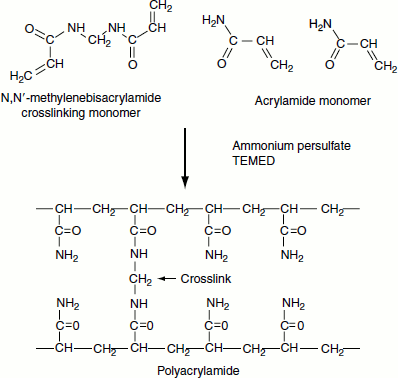
Figure 12.5 Stacking and separating gel
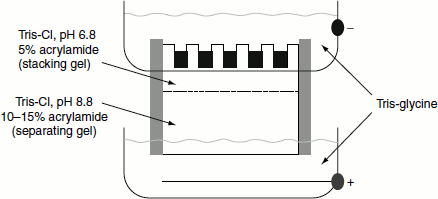
Stacking gel: This region of the gel has high pore size; usually, the stacking gels are about 5 per cent. The function of the stacking gel is to concentrate the protein samples on the separating gel. They are prepared using tris-HCl buffer of pH 6.8.
After the separating gel sets, the stacking gel solution is poured and allowed to set after placing the comb. Once the gel is set, the comb can be removed to create sample wells.
Sample loading: The gel plate is inserted into the electrophoretic unit and the reservoirs are filled with the running buffer. The comb is then removed after clearly marking the position of the wells on the gel plate.
The sample is then solubilized in the sample solubilizing buffer (SSB), which is a mixture of SDS, buffer, β-mercaptoethanol, glycerol and the tracking dye bromophenol blue. The protein sample treated with SSB is then loaded to the wells.
Electrophoretic running: Once the sample is loaded to the gel, the electrophoretic process is started by connecting the unit to power pack. Initially, the voltage is set at minimum (50 V) till the time the sample is in the stacking gel region. The mobility of the sample can be visualized as a blue line, which is the tracking dye, bromophenol blue. After the sample crosses the stacking gel, the voltage is increased (100 V). Usually, electrophoresis is carried out in air-conditioned rooms. This is to minimize the heat developed during the process.
Electrophoretic mobility: The stacking gel contains chloride ions, the leading ions, which migrate more quickly through the gel than the protein sample, while the electrophoresis buffer contains gly-cine ions, the trailing ions, which migrate more slowly. The protein molecules are trapped in a sharp band between these ions. As the protein enters the separating gel, which has a smaller pore size, a higher pH and a higher salt concentration, the glycine is ionized, the voltage gradient is dissipated and the protein is separated based on size.
- In stacking gel, pH 6.8:

- Negatively charged form Gly is disfavoured.
- Average electrophoretic mobility is very low.
- In separating gel, pH 8.8

- Negatively charged form is favoured.
- Average electrophoretic mobility much higher.
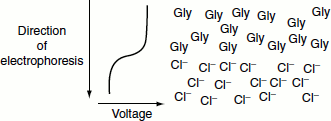
- If a Gly molecule diffuses ahead into Cl– region, it experiences a lower voltage and slows down. If a Cl–ion diffuses back into the Gly region, it experiences a higher voltage and speeds up until it reaches the boundary.
- Ion boundary and voltage gradient become progressively shaper.
- Proteins are trapped between Gly and Cl–.
- Proteins form a very tight band.
In the separating gel
- pH is increased to 8.8.
- Negatively charged form of Gly is favoured.
 Glycine mobility increases; it becomes greater than protein mobility, but still slower than Cl–.
Glycine mobility increases; it becomes greater than protein mobility, but still slower than Cl–. 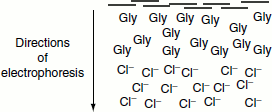
- Ion front moves ahead of proteins.
Protein sample, now in a narrow band, encounters both the increase in pH and the decrease in pore size. The increase in pH would tend to increase the electrophoretic mobility; however, smaller pores decrease mobility. The relative rate of the movement of ions in lower gel is chloride > glycinate > protein. Proteins separate based on charge/mass ratio and on size and shape parameters.
Visualization of the gel: The separated proteins on the gel can be visualized after staining and distaining the gel. The staining solution is a mixture of Coomassie Brilliant Blue stain (CBB), 50 per cent methanol and 10 per cent acetic acid. After electrophoresis, the gel is soaked in the staining solution for 1–2 h. The gel is then transferred to the destaining solution which is a mixture of 50 per cent methanol and 10 per cent acetic acid. The gel is destained until it gives an appropriate band.

Figure 12.6 SDS – PAGE electrophoretic pattern