Cells all of which contain the same DNA sequences are called ‘clones’. Cloning serves two main purposes.
- From a limited number of starting material, a large number of rDNA molecules can be produced.
- A second important function is purification. No other DNA molecules are present at the end of the procedure.
The various steps involved in molecular cloning can be outlined in the following sections.
Preparation of Vector DNA
Vector DNAs that originate from microorganisms are propagated and harvested from their appropriate microbial hosts. The bacterial cells grown in a nutrient broth are harvested by low-speed centrifugation (5,000 g for 20 minutes) at 4°C. After the supernatant is discarded, a large bacterial pellet is left. In order to release the plasmid DNA from this pellet, the cells must be ruptured. This can be achieved by a variety of techniques. Sonication and boiling can be used but the most common method is using lysozyme. Lysozyme degrades the peptidoglycan cell wall of bacteria very efficiently. After the cell wall has been disrupted and cell lysis occurs, the plasmid DNA needs to be separated from the cellular proteins and the high molecular weight chromosomal DNA. This can be achieved by treating the lysed cells with the detergent sodium deodecylsulphate (SDS) (1 per cent W/V) and 0.2-M sodium hydroxide. This SDS/NaOH treatment denatures the double-stranded DNA and solubilizes the protein. The alkali treatment is followed by the acid treatment with 3-M sodium acetate in a pH of 4.5. After incubation on ice for an hour, the mixture is centrifuged at high speed (17,000 g) for one hour. This procedure causes acid precipitation of the protein and high molecular weight chromosomal DNA, which are pelleted, leaving the plasmid DNA in the supernatant. The supernatant can then be treated with ethanol and incubated at 0°C-20°C in order to precipitate the plasmid DNA. The plasmid DNA can then be pelleted by centrifugation.
Preparation of Target DNA
The tissue containing the DNA of interest is crushed with a homogenizer. The macerated tissue is placed in a digestion buffer (100-mM NaCl; 0.1 mg/ml proteinase K enzyme). The tissue is digested in the presence of a detergent at 50°C for 12–24 hours. Following the digestion step, the mixture is treated with the organic mixture of phenol and chloroform in order to extract the chromosomal DNA. The DNA is retained in the aqueous phase and the proteins are denatured and extracted into the phenol chloroform organic phase. The DNA is then treated with ethanol that precipitates the DNA and pelleted by centrifugation. The pellet is then washed in 70 per cent ethanol, centrifuged and resuspended in suitable buffer.
Construction of rDNA
The vector and the target DNA are cut with appropriate restriction enzyme and are then ligated together by DNA ligase.
Transport into the Host Cell (Transfection)
There are different ways of transporting the rDNA into a host cell. Some of the methods include:
- Using calcium chloride,
- Electroporation,
- Nucleofection,
- Liposome-mediated gene transfer,
- Particle bombardment and
- Microinjection.
Using calcium chloride
A solution of 50-mM CaCl2causes the rDNA to precipitate onto the outside of bacterial cells (host). Then, a brief heat shock is given by raising the temperature to 45°C. Calcium ions promote cell membrane lysis that facilitates DNA transfer into the cell, subsequent heat shock also promotes the process.

Electroporation
This method employs the use of high-voltage electric pulse to introduce the rDNA into the host cell. The host cells are subjected to an electric pulse of 2,500 V for 3–5 m/sec. Some host cells are killed by this process; however, many survive and take up the rDNA.
Nucleofection
Nucleofection is the method of introducing DNA molecules efficiently into the nucleus of virtually any cell type, therefore, significantly increasing the chances of chromosomal integration of the transgene.
Liposome-mediated gene transfer
Polycationic lipid and a neutral lipid when mixed will result in the formation of unilamellar liposome vesicles that have a net positive charge due to the highly positive amine head groups on these molecules. Liposome-mediated gene delivery is technically easy, highly reproducible and very efficient.
Particle bombardment
This is a valuable tool for molecular biologists and permits direct gene transfer to a wide range of cell and tissue types. Some of the important applications of the process include the production of transgenic crop species including maize and soya bean and the introduction of DNA into plastids and mitochondria.
Microinjection
The host cell is immobilized by applying a mild suction with a blunt pipette. The foreign gene is then injected with a microinjection needle. Transgenic mice have routinely been generated by injecting a solution of DNA into a pronucleus of a fertilized egg. The DNA becomes integrated into one of the chromosomes. Cells descending from this fertilized egg, including germ cells, can contain the transgene DNA/rDNA (Figure 9.27).
Figure 9.27 Microinjection

Selection of Transformed Cells
The ligation of vector DNA with the DNA of interest may result in:
- Unligated vectors,
- Recircularized vector molecules without insert and
- Host cells that had not taken up the rDNA.
Therefore, the selection of the recombinant host cells is important. This can be done by the following methods:
- Insertional inactivation of selectable marker gene.
- Insertional inactivation of antibiotic-resistant gene.
PBR322 plasmid has ampr and tetr genes. For Bam H I, there is a unique restriction site within the tetr gene. Therefore, if the DNA of interest is inserted and the plasmid vector is transformed into E. coli, the amps and tets E. coli hosts become ampr and tets. The various steps involved can be discussed as follows:
- E. coli cells are transformed with the recombinant plasmid.
- The E. coli cells are then plated on ampicillin-containing agar. All the colonies that grow in this medium will be transformed cells, as untransformed cells cannot grow in this medium.
- The bacterial culture is then ‘replica plated’ onto an agar containing tetracycline. In this medium, only ampr and tetr cells will grow, i.e., recombinants will not grow in this medium.
- The recombinants can be picked from the master plate.
Insertional inactivation of other gene
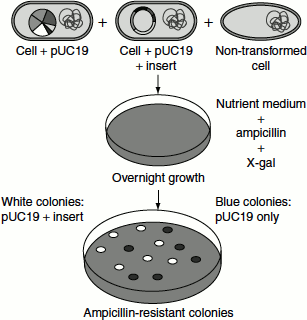
pUC8 plasmid carries ampr genes and lacZ’ genes. The lacZ’ gene codes for the α-peptide portion of the enzyme β-galactosidase. When this plasmid is inserted into an E. coli that lacks lacZ’ segment (i.e., lacZ’ mutant), the bacterial and plasmid genes complement each other to produce a functional β-galactosidase. This process is called ‘α-complementation’ (Figure 9.28). If a foreign gene is inserted into this lacZ’ gene of pUC plasmids, then it cannot complement and no functional β-galactosidase is formed, i.e., recombinants show no β-galactosidase activity.
Figure 9.28 Selection of recombinants by a-complementation

The various steps involved are:
- Recombinants are first selected by growing in ampicillin-containing medium.
- E. coli cells with normalpUC8 plasmid will be ampr and can synthesize β-galactosidase.
- Recombinants will be ampr but cannot synthesize β-galactosidase.
- When X-gal (5-bromo-4-chloro-3-indoyl-β-D-galactopyranoside) is added to the medium, the recombinant colo-nies will appear white in colour, as they do not have β-galactosidase; while non-recombinants will appear blue in colour, as β-galactosidase breaks X-gal to give a blue colour (Figure 9.29).
Figure 9.29 Recombinant selection using X-gal