Chromosomal regions that are activated for transcription are marked by a variety of structural changes. The packaging of eukaryotic DNA into chromatin limits its availability as a template for transcription. Thus, modifying chromatin structure plays a key role in controlling the gene expression in eukaryotes. The transcription of eukaryotic gene is strongly repressed when it is condensed as chromatin. The presence of histone protein and nucleosome complexes limits the availability of the DNA sequences for the binding of transcription factors and RNA polymerase. Therefore, the decondensation of the chromatin and the disruption of the nucleosome structure is the key control point of gene regulation. A series of transcription-associated changes takes place in the chromatin and this is referred to as ‘chromatin remodelling’. Once established, such changes in chromatin can persist through cell divisions, creating an epigenetic state in which properties of the gene are determined by the self-perpetuating structure of the chromatin.
The DNA of all eukaryotic cells is tightly bound to histone proteins. The basic structural unit of chromatin is the nucleosome, which consists of 146 bp of DNA wrapped around two molecules each of histone H2A, H2B, H3 and H4, with one molecule of histone H1 bound to the DNA, as it enters the nucleosome core particle. The chromatin is then condensed by being coiled into higher-order structures (refer Chapter 2).
Chromatin Remodelling
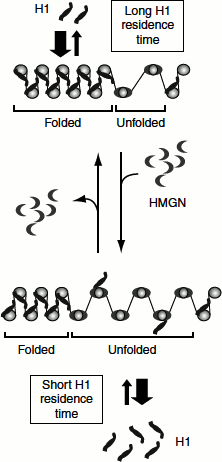
Chromatin remodelling accompanying transcription initiation involves the modifications of histones, the rearrangements of nucleosomes and the association of various non-histone proteins such as HMGN proteins (high mobility group nucleosome-binding protein). The binding site of HMGN proteins on the nucleosome overlaps the binding site of histone H1 (Figure 7.8).
HMGN proteins, therefore, compete with H1 for shared binding sites on the nucleosome, decrease the H1 residence time at selected chromatin loci and promote the unfolding of the chromatin fibre.
Chromatin remodelling is performed by ATP-dependent chromatin-remodelling complexes, which use the energy of ATP hydrolysis for remodelling. The core region of the chromatin-remodelling complex is its ATPase subunit. The remodelling complexes are classified according to the subfamilies of ATPase that they contain as their catalytic sub-unit. There are many sub-families, the four major ones are SWI/SNF(switch sniff complex), ISWI, CHD and INO80/SWR1. SWI/SNF can remodel in vitro without the loss of histones or can displace histone octamers. The structure of the target nucleosome is altered leading to a remodelled nucleosome on the original DNA or may displace the histone octamer to different position on the DNA. The SWI/SNF complexes generally are involved in transcription activation.
The ISWI family primarily affects the nucleosome positioning without displacing octamers, in a sliding reaction in which the octamers moves along the DNA. The ISWI complexes act as repressors of transcription.
Figure 7.8 Chromatin remodelling by HMGN proteins
Nucleosome-Remodelling Factors
These are protein complexes that alter the arrangement of nucleosomes, without removing or covalently modifying histones. One mechanism by which they act is to catalyse the sliding of histone octamers along the DNA molecule, thereby repositioning nuceleosomes to change the accessibility of specific DNA sequences to interact with transcriptional regulatory proteins.
Histone Modifications
All of the core histones are subject to multiple covalent modifications. Different modifications result in different functional outcomes. The modifications include acetylation, methylation and phosphorylation.
Histone acetylation
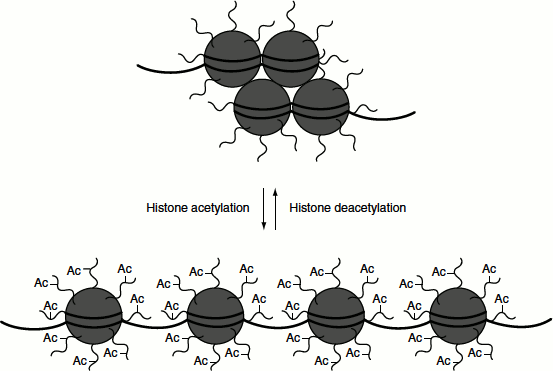
This is often correlated with transcriptionally active gene in a wide variety of cell types. The core histones H2A, H2B, H3 and H4 have two domains: a ‘histone fold domain’, which is involved in interactions with other histones and in wrapping DNA around the nucleosome core particle and an ‘amino terminal histone tail domain’, which extends outside of the nucleosome. Amino terminal histone tail domain is rich in lysine residues. All core histones are acetylated on lysine residues. Acetylation reduces the net positive charge of the histones and may weaken their binding to DNA as well as alters their interaction with other proteins. In addition, the acetylation of histones facilitates the binding of transcription factors to nucleosomal DNA. Thus, histone acetylation increases the accessibility of chromatin to DNA-binding proteins. The enzymes involved for the acetylation of lysine residues are ‘lysine (K) acetyltransferases or KATs’; when they specifically acetylate the lysine residues of histone, they are referred to as histone acetyl transferase (HAT). There are two classes of HATs namely group A and group B. Group A acts on histones in chromatin and is involved in transcription control. While group B acts on newly synthesized histones in the cytosol. The acetylation reactions are reversible and the acetyl groups are removed by histone deacetylases (HDACs). The deactylation of histones is linked with transcription repression. The absence of histone acetylation is a feature of heterochromatin. Active chromatin is acetylated on tails of histone H3 and H4. Inactive chromatin is methylated on a specific lysine of histone H3 (Figure 7.9).
Histone methylation
Histone methylation can be linked with either active or inactive regions depending on the specific sites of methylation. Histones H3 and H4 are methylated at lysine residues in the tail. In addition, three arginines in H3 and H4 are also methylated. Trimethylated H3K4 occurs at the transcriptional start site of active genes. H3 methylated at K9 or K27 is a characteristic feature of transcriptionally silent genes. Histone methylations are characterized by lysine methyltransferases (KMTs or HMTs). Methylation is also reversible and can be demethylated by lysine demethylases.
Histone phosphorylation
All histones can be phosphorylated in different contexts inside the cell. Histone phosphorylation is commonly seen in three different circumstances.
- During the cell cycle,
- Chromatin remodelling during transcription and
- During DNA repair.
Figure 7.9 Histone acetylation
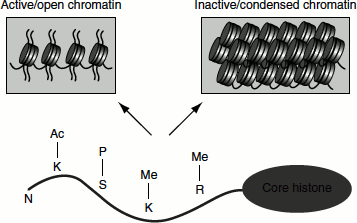
Histone H1 is phosphorylated at mitosis. The phosphorylation of serine 10 of histone H3 is linked to transcriptional activation (where it promotes the acetylation of K14 in the same tail), as well as to chromosome condensation and to mitotic progression. It is to be noted that histone phosphorylation during cell division favours chromatin condensation; however, during transcription and DNA repair, it favours chromatin decondesation (Figure 7.10).
Histone-modifying enzymes, in particular HATs and HDACs, have been shown to be involved in the generation of cancer and other diseases such as Rubinstein–Taybi syndrome (RTS), a mental disorder accompanied by skeletal abnormalities, acute myeloid leukaemia (AML) and certain gastric and colorectal cancers. The amplification and overexpression of another HAT correlates with breast cancer. HDACs are also implicated in cancer, for example, promyelotic leukaemias.
Figure 7.10 Histone modifications modulate chromatin structure
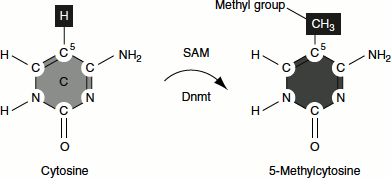
DNA Methylation
The methylation of DNA is another mechanism by which the control of transcription is linked to chromatin structure. Cytosine residues in vertebrate DNA can be modified by the addition of methyl groups at the fifth carbon position. DNA is methylated at the Cs that precedes the Gs in the DNA chain (CpG dinucleotide). This methylation is correlated with the reduced transcriptional activity of genes. High frequencies of C, G nucleotides near the promoter sequences reduce gene expression. Methylation inhibits the transcription of these genes by interfering with the binding of some transcriptional activators, as well as recruiting repressors that specifically bind methylated DNA. The methylation pattern is heritable after cell division. Therefore, DNA methylation plays an important role in cell differentiation during development. ‘Epigenetics’ is the study of heritable changes in chromatin (e.g., DNA methylation) without involving the change in DNA sequences.